Recent Development on Amendments of the
Drug Administration Law
By Manuel Torres (Managing Partner of Garrigues China) and Cindy Pan (Senior Associate)
 就在去年10月,国家食品药品监督管理总局(CFDA)发布了“《中华人民共和国药品管理法》修订版草案(草案征求意见稿)”(下文简称“修订草案”),拟取消GMP/GSP认证,实行药品上市许可持有人制度。由药品上市许可持有人,对药品安全、有效和质量可控承担法律责任。
就在去年10月,国家食品药品监督管理总局(CFDA)发布了“《中华人民共和国药品管理法》修订版草案(草案征求意见稿)”(下文简称“修订草案”),拟取消GMP/GSP认证,实行药品上市许可持有人制度。由药品上市许可持有人,对药品安全、有效和质量可控承担法律责任。
修正案草案征求意见稿对现行《药品管理法》增加6条,修改9条,删去2条。主要修改内容包括:
1. 全面实施药品上市许可持有人制度。草案征求意见稿总结了药品上市许可持有人制度试点经验,全面落实药品上市许可持有人制度。明确取得药品批准文号的申请人,为药品上市许可持有人。药品上市许可持有人可以自行生产经营药品,也可以委托他人生产经营。药品上市许可持有人对药品临床前研究、临床试验、生产经营、不良反应报告等承担全部法律责任。
2. 取消GMP、GSP认证。认证被取消后,取而代之的是“全面实施药品上市许可持有人制度”,上市许可持有人将对药品安全、有效和质量可控承担法律责任。
3. 落实行政审批制度改革要求。将临床试验机构由认证改为备案,药物临床试验审批由明示许可改为默示许可,生物等效性试验实行备案管理。取消药品生产质量管理规范认证、药品经营质量管理规范认证制度。将原料药和辅料修改为与药品一并审批。
4. 落实处罚到人要求。对存在资料数据造假和被吊销许可证的单位及其直接负责的主管人员和其他直接责任人员,十年内行业禁入;因药品安全犯罪被判处有期徒刑以上刑罚的人员,终身不得从事药品的研制、生产、经营、进出口活动。药品上市许可持有人、研制单位、生产企业、经营企业、医疗机构故意实施违法行为,存在重大过失、违法行为情节严重、性质恶劣或造成严重后果以及其他严重不良社会影响的,对单位直接负责的主管人员和其他直接责任人员处以其上一年度从本单位取得的收入百分之三十以上一倍以下的罚款。
为确保《创新意见》相关改革措施尽快实施,此次《药品管理法》只是局部修改,对法律篇章结构调整、药品定义和分类、药品全链条和全生命周期管理、监管措施细化等暂未涉及;专利链接、专利期补偿等探索和试点工作暂不列入,将按程序报请全国人大常委会授权。下一步,食品药品监管总局将加快《药品管理法》全面修订工作进程,上述内容将在修订草案中予以充分体现。
 To deepen the reform of drug evaluation and approval system, encourage innovations in drugs, and safeguard rights and interests of the public with regard to drug consumption, China Food and Drug Administration (the “CFDA”) has published series of pharmaceutical laws and regulations recently. Among others, the most notable is the Drug Administration Law of the People’s Republic of China (draft for consultation) (the “Draft”) promulgated on October 23rd, 2017, due to several major reforms in the area of drug registration and administration.
To deepen the reform of drug evaluation and approval system, encourage innovations in drugs, and safeguard rights and interests of the public with regard to drug consumption, China Food and Drug Administration (the “CFDA”) has published series of pharmaceutical laws and regulations recently. Among others, the most notable is the Drug Administration Law of the People’s Republic of China (draft for consultation) (the “Draft”) promulgated on October 23rd, 2017, due to several major reforms in the area of drug registration and administration.
According to the Draft, China will fully adopt a drug Market Authorization Holder (the “MAH”) system, the approval regime of clinical trials will be simplified, and overseas trial data will be accepted in drug registration, though an on-site check by CFDA might still be applied.
1. Implementing Drug Marketing Authorization Holder System
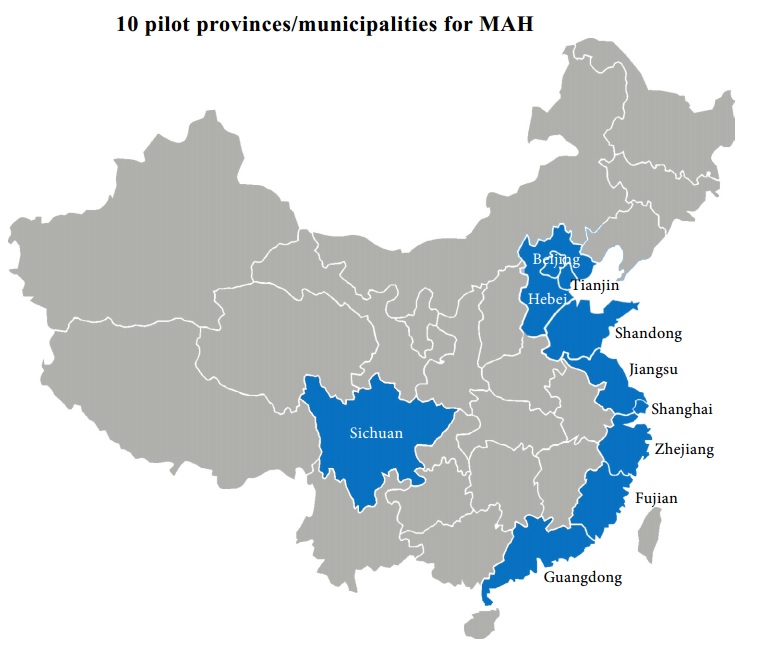
As early as in 2015, the document Guo Fa [2015] No.44 (Decision of the Standing Committee of the National People's Congress on Authorizing the State Council to Carry out the Pilot Drug Marketing License Holder System in Certain Places and Relevant Issues) proposed to launch a pilot MAH scheme. In 2017, the State Council issued an effective notice (the Circular of the General Office of the State Council on Promulgating the Pilot Program for the Drug Marketing Authorization Holder System (Guo Ban Fa [2016]No.41) (the “Pilot Program”)), dated May 26th, 2016, formally authorizing a trial plan for new drug MAH system in ten provinces: Beijing, Shanghai, Tianjin, Hebei, Jiangsu, Zhejiang, Fujian, Shandong, Guangdong, and Sichuan. Pharmaceutical research institutions and individual researchers in these provinces can submit an application for clinical trials or marketing authorization registration. Applicants obtaining marketing authorizations and approval documents can become MAHs and take legal responsibility for clinical trials, production, and marketing, which was previously not allowed.
According to the Draft, a new article is introduced as Article 5, reading “the State runs the drug marketing authorization holder system, and parties authorized to launch drugs on the market shall bear legal liability for the controllable safety, effectiveness and quality of drugs”, which means that the MAH system will be rolled out nationwide soon.
 (1) Qualification of Marketing Authorization Holder
(1) Qualification of Marketing Authorization Holder
Article 31 of the Draft provides that "a MAH shall be an applicant that succeeds in obtaining a drug approval number", which does not clearly state the scope of subject for being an applicant.
According to the Pilot Program for the System of MAH issued in 2016, applicants include all drug research institutes in the pilot administrative region and researchers working in the pilot administrative region concerned with the nationality of the People's Republic of China. However, in accordance with the Measures for the Administration of Drug Registration (revised version) promulgated on the same day of the Draft, there should be a requirement that the domestic applicant for the registration of the drug shall be a pharmaceutical manufacturer or Research & Development institution (the “R&D institution”) in China and that the overseas applicant should be foreign legal pharmaceutical manufacturers. Therefore, if the Draft is implemented simultaneously with the revised Administration of Drug Registration, natural persons may not be able to become MAH.
(2) Separation of Drug Marketing Approval and Drug Manufacturing Approval
For a long time, China has been implementing the system of binding drug manufacturing approval and drug marketing approval according to current Drug Administration Law . Under this system, R&D institutions may obtain new drug certificates. However, if they want to put the drugs into production, they must invest a lot of money into self-built factories and production lines to obtain drug manufacturing licenses, GMP certificate, in the end, applying for drug approval numbers. Due to lack of capital for R&D institutions, therefore, most of them are more inclined to transfer their R&D achievements to pharmaceutical manufacturers in the form of technology transfer. For R&D institutions, the cost of self-built is too expensive, and the transfer of technology may lead to a low return rate on R&D investment. Both approaches are not the best choice.
Now, the Draft has made a major improvement of this system. In accordance with Article 31 of the Draft “Where a drug marketing authorization holder manufactures drugs on its own, it shall have obtained a drug manufacturing certificate; where it distributes drugs itself, it shall meet the requirements on drug distribution as stipulated in this Law; where it entrusts a drug manufacturing or distributing enterprise with the required qualifications to manufacture or distribute its drugs, it shall enter into an entrustment agreement with the entrusted enterprise to clarify rights, obligations and duties of each party, in order to ensure that the manufacturing or distribution activities of the entrusted enterprise comply with the requirements set out in this Law”, this article 31 formally separates the listing of drugs from the manufacture approval, which encourages R&D institutions. In the meantime it is also good news for those without research and development capabilities’ in pharmaceutical factories.
 (3) Change of MAHs
(3) Change of MAHs
All along, there are no laws and regulations on the issue whether the MAHs can be changed or transferred, which urgently needs a solution due to the actual needs for transfer, inheritance, M&A and dissolution in practice.
In the Draft, it officially said that “Changing the drug MAH requires the satisfaction of the requirements set forth in this Law and shall be subject to the approval of the drug regulatory department under the State Council”, which means changing or transferring the MAH is legally feasible in future.
This improvement has brought us in line with international standards, establishes the long-term development trend of the MAH system, and promotes the development of Chinese medical science.
(4) Obligation of Re-evaluation of MAH
The current Drug Administration Law stipulates that the drug regulatory department under the State Council shall re-evaluate the drugs that have been approved for manufacturing and marketing . However, the Draft attributed the drug re-evaluation obligations in Article 34 to the MAHs and has further stipulated in the same article that “Where a drug MAH fails to perform its obligations of re-evaluating drugs as required, the drug regulatory department under the State Council shall order it to carry out such re-evaluation. The said department may directly organize the reevaluation of drugs in question when necessary”.
Although the Draft does not directly impose strict legal liabilities on the MAH for not performing drug re-evaluation obligations, MAH should pay adequate attention.
(5) Legal Liabilities of MAH
The Draft sets clear legal liability for the MAH. Article 94 of the Draft stipulates that “any MAH that violates the provisions of Article 32 in this Law shall be fined more than CNY 100,000; where the case is serious, it shall be ordered to suspend production or business operation for rectification, with its drug approval document withdrawn as the worst measure taken; where a crime is constituted, its criminal liability shall be investigated.”
In addition to the aforementioned legal liability clause of Article 94, the Draft also adds corresponding liabilities in the relevant legal obligations and liabilities provisions of the MAH, e.g. Article 71 , 72 , 80 . At the same time change the “drug manufacturer” to “MAH” in the relevant provisions, e.g. Article 90 , 91 , 93 .
It is noteworthy that although changes are reasonable in the relevant clauses mentioned above, the manufacturing factories which are only authorized for production are likely to evade the relevant legal liability on this ground.
At present, Shanghai, Fujian and other pilot cities have issued specific guidelines and material requirements for applying for the MAH. Once the Draft is implemented, the system of the MAH will greatly enhance the innovation initiative of drug R&D institutions, reduce the cost of R&D investment, release the production capacity and promote innovation and development of pharmaceuticals.
2. Cancelling Certificates of GMP and GSP
 Aticle 10 of the Draft has cancelled the requirement that drug manufacturing enterprises shall acquire the certificate of GMP and modifies to "Drug manufacturers shall comply with the Good Manufacturing Practice for Drugs formulated by the drug regulatory department under the State Council based on this Law, establish and improve their own quality management system, and ensure compliance at all times during production processes".
Aticle 10 of the Draft has cancelled the requirement that drug manufacturing enterprises shall acquire the certificate of GMP and modifies to "Drug manufacturers shall comply with the Good Manufacturing Practice for Drugs formulated by the drug regulatory department under the State Council based on this Law, establish and improve their own quality management system, and ensure compliance at all times during production processes".
Meanwhile, Article 16 of the Draft has cancelled the requirement that drug supply enterprise shall acquire the certificate of GSP, while it changes to "Drug distributors must deal in drugs in accordance with the Good Distribution Practice for Drugs formulated by the drug regulatory department under the State Council based on this Law".
Cancellation of GMP and GSP certification actually reflects the changes in regulatory thinking of drug regulatory authorities from "certificate-oriented" to "regulatory-oriented". Simplification of administrative examination and approval has reduced administrative procedures and has saved administrative resources. However, legal liabilities of pharmaceutical manufacturers and distributors have not been lightened by the simplification. In accordance with the Article 79 of the Draft, “any drug manufacturer, drug distributor, institution for non-clinical safety study, institution for drug clinical trial or contractual research institution that does not implement the Good Manufacturing Practice for Drugs, the Good Distribution Practice for Drugs, good practices of non-clinical drug research or good practices of clinical drug trials as required shall be given a warning and ordered to make corrections within a time limit.
If it fails to do so within the time limit, it shall be ordered to suspend production or business operation for rectification and shall concurrently be fined not less than CNY 5,000 but not more than CNY 20,000. If the circumstances are serious, the Drug Manufacturing Certificate or Drug Distribution Certificate shall be revoked, and the institution for non-clinical safety study, institution for drug clinical trial, or contractual research institution concerned shall be prohibited from carrying out non-clinical safety studies of drugs or clinical trials of drugs for five years. Where a crime is constituted, criminal liability shall be investigated”. It can be concluded that it puts higher and more stringent requirements to the pharmaceutical manufacturers and distributors.
3. Simplification of Administrative Approval of Clinical Trials
(1) Admittance to Drug Clinical Trail Institutions from Qualification to Record-filing
Another important reform in the Draft is reflected in Article 29 with respect to clinical trial of drug, which states that “Clinical trials shall be carried out in clinical trial institutions with necessary infrastructure. Clinical trial institutions shall be subject to administration by record-filing, and specific measures for this purpose will be formulated by the drug regulatory department under the State Council and the health administration under the State Council together”. According to the current Drug Administration Law and the Criterions for the Quality Control of Clinical Trial of Drugs, drug clinical trial agencies shall be qualified and accredited by the relevant authority.
With respect to legal liability, Article 79 of the Draft states that clinical trial institutions do not conform to Good Clinical Practice ("GCP") will be served a warning and ordered to correct within the prescribed time limit, as well as “where an institution for drug clinical trial fails to file a record as required under Article 29 of this Law or a bioequivalence test is conducted without filing a record as required under Article 29 of this Law, the party concerned shall be ordered to make corrections and be warned, and may also be fined less than CNY 100,000”.
The draft for comment version of specific regulation Administrative Provisions on Drug Clinical Trial Institutions has been published by CFDA on 26th October, 2017.
For this reform, CFDA officially said that record-filing is not to lower the access standards. On the contrary, this measure has actually raised the standard. Each filled clinical trial institution will receive a record-filing number, and the relevant authority will conduct routine supervision on these institutions.
 (2) Changes of Clinical Trial Approval from Express Permission to Implied Consent
(2) Changes of Clinical Trial Approval from Express Permission to Implied Consent
The Draft stipulates the approval of clinical trial in Article 29 that “the drug regulatory department under the State Council shall decide whether to agree on the clinical trials or not within 60 working days from the date on which an application for proceeding to clinical trials has been accepted; the applicant may carry out clinical trials if the said department fails to issue a notification within the required time limits”. The reform posed new challenges to the ethics committee, under which the ethics committee becomes a prerequisite for approval of clinical trials according to Article 29 .
In addition, what requires special attention recently is that CFDA released a decision on Adjusting Relevant Matters Concerning the Administration of Imported Drug Registration (the “Decision”). According to the Decision, in case of a drug subject to an international multi-center clinical trial ("IMCCT") of drugs to be conducted in China, the phase I clinical trial of the drug is allowed simultaneously, and the requirement on the drug subject to the clinical trial to have been registered overseas or have entered a phase II or III clinical trial shall be cancelled, except for biological products for prevention. Upon completion of the IMCCT of a drug in China, an applicant may directly file a registration application for marketing. For a new chemical drug or innovative therapeutic biological drug that is applied for clinical trial and marketing as an imported drug, the requirement on the drug to have obtained a marketing license issued by the country or region where the drug's overseas pharmaceutical manufacturer is located shall be removed. This improvement has greatly accelerated the progress of registration of imported drugs as well as to facilitate Chinese parties’ early participation in IMCCT.
These are major moves by the CFDA to come more close to international standards since officially joining the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) in 2017.
 4. Definitude and Concretization of Legal Liabilities
4. Definitude and Concretization of Legal Liabilities
Comparing the current Drug Administration Law, the Draft adds legal liabilities of CRO according to Article 79 in order to comply with good practices of non-clinical drug research or good practices of clinical drug trials as required.
The Draft emphasizes the legal responsibility of non-clinical safety evaluation research institutes, drug clinical trial institutes and contract research organizations in case of data infringement in Article 95 , as well as the direct imposition against chief directly in-charge and other personnel directly held liable Article 96 .
 5. Conclusion
5. Conclusion
There is no doubt that this modification is a significant improvement in the reform of drug regulatory regime in China and it is in line with international standards, but it also faces numerous challenges. Overall, the drug R&D institutions, distributers and manufacturers will be greatly benefited from this reform and it will optimize resource allocation.

